# Overview 
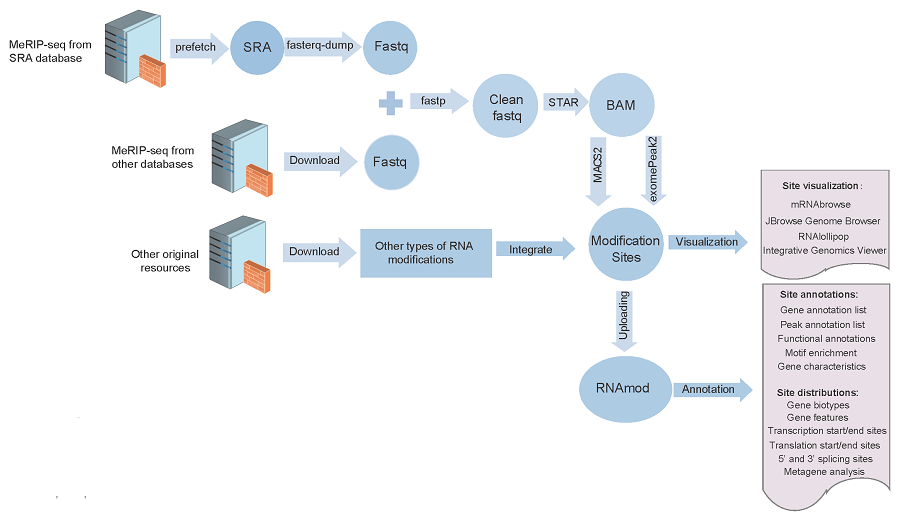
The MeRIP-seq datasets in the SRA format were converted to the FASTQ format using sratoolkit and then the default parameters of fastp were used to trim the adapter sequences and low-quality bases. After filtering the data for quality, STAR was used to map clean reads to the corresponding reference genomes to generate BAM format results. And then using the BAM format genome mapping files of the IP and the input samples, we applied two peak calling strategies. The following parameters of MACS2 were used to identify methylation peaks: --nomodel --extsize 150 -B -n -q 0.05. The R package exomePeak2 (https://github.com/ZW-xjtlu/exomePeak2) was used for peak calling, which involved the same BAM files and GTF files. The parameters were set as follows: fragment_length = 100 binding_length = 25 step_length = 25 pc_count_cutoff = 5 bg_count_cutoff = 50 p_cutoff = 1e-05 peak_calling_mode = exon.
RNAmod (https://bioinformatics.sc.cn/RNAmod), which is an interactive and freely available platform for the annotation and visualization of RNA modifications, was used to annotate the RNA modifications in PRMD. First, RNAmod extracted gene features, such as promoter regions, 5′ and 3′ untranslated regions (UTRs), start codon regions, coding sequence (CDS) regions and stop codon regions, from different annotated reference genomes and then examined gene characteristics, including the GC content, length and minimum free energy. Second, RNAmod mapped all modification sites to different RNA features and calculated coverage values and analyzed metagenes and other annotations. The modified genes were functionally characterized on the basis of Gene Ontology (GO) and KEGG pathways using the clusterProfiler package and according to Reactome pathways using ReactomePA packages.
# The Analysis Pipeline of Peak Calling in Different Plant Species
# Building work environment
The software needs to install:
sratoolkit.2, STAR,fastp,samtools,MACS2,R 4.2,exomePeak2 (R package)
Example files and Scripts:
| Example files |
Download link |
| id.txt |
|
| sra2srx_id.txt |
|
| srrid.txt |
|
| Scripts |
Download link |
| download.sh |
|
| exomePeak2.r |
|
| get_bam.sh |
|
| macs2.sh |
|
| rename_bam_file.sh |
|
Database:
### take Arabidopsis thaliana as an example
## reference sequence download
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/fasta/arabidopsis_thaliana/cds/Arabidopsis_thaliana.TAIR10.cds.all.fa.gz
## or deliver tasks to the background server
nohup wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/fasta/arabidopsis_thaliana/cds/Arabidopsis_thaliana.TAIR10.cds.all.fa.gz &
## GTF format annotation file download
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.57.gtf.gz
or
nohup wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.57.gtf.gz &
## MeRIP-seq datasets download
#srrid need to be prepared
# cat download.sh
#!/usr/bin/bash
cat srrid|while read id
do
$software_path/sratoolkit.2.11.0-centos_linux64/bin/prefetch $id
Done
# run download.sh
nohup sh download.sh software_path &
# Get BAM formatted files
### run get_bam.sh to get BAM formatted files
sh get_bam.sh species dir_path soft_path
### rename the bam files
##Convert srrid_name bam file to srxid_name bam file, the file sra2srx_id is needed, here take 12 MeRIP-seq samples of Arabidopsis thaliana as an example
## cat sra2srx_id.txt
SRR13522944 SRX9933631
SRR13522945 SRX9933630
SRR13522946 SRX9933629
SRR13522947 SRX9933628
SRR13522948 SRX9933627
SRR13522949 SRX9933626
SRR13522950 SRX9933625
SRR13522951 SRX9933624
SRR13522952 SRX9933623
SRR13522953 SRX9933622
SRR13522954 SRX9933621
SRR13522955 SRX9933620
## get rename_bam_file.sh
awk '{print "mv""\t"$1"Aligned.sortedByCoord.out.bam""\t"$2".bam"}' sra2srx_id >>rename_bam_file.sh
## run rename_bam_file.sh
sh rename_bam_file.sh
# Peak calling use MACS2
### take 12 MeRIP-seq samples of Arabidopsis thaliana as an example
## cat id.txt
IP Input
SRX9933630 SRX9933620
SRX9933631 SRX9933626
SRX9933621 SRX9933629
SRX9933625 SRX9933622
SRX9933627 SRX9933623
SRX9933628 SRX9933624
## cat macs2.sh
#!/usr/bin/bash
software_path=$1
inputs=(SRX9933620 SRX9933626 SRX9933629 SRX9933622 SRX9933623 SRX9933624)
ips=(SRX9933630 SRX9933631 SRX9933621 SRX9933625 SRX9933627 SRX9933628)
for(( i=0;i<${#inputs[@]};i++)) do
inputSample=${inputs[$i]}.bam
ipSample=${ips[$i]}.bam
$software_path/macs2 callpeak -t $ipSample -c $inputSample -f BAM --nomodel --extsize 150 --outdir macs2_out -B -n ${ips[$i]}_${inputs[$i]} -q 0.05
done
## run macs2.sh
nohup sh macs2.sh oftware_path &
# Peak calling use exomePeak2
### take 12 MeRIP-seq samples of Arabidopsis thaliana as an example
## cat id.txt
IP Input
SRX9933630 SRX9933620
SRX9933631 SRX9933626
SRX9933621 SRX9933629
SRX9933625 SRX9933622
SRX9933627 SRX9933623
SRX9933628 SRX9933624
## cat exomePeak2.r
library("exomePeak2")
sample_list <- read.csv("id.txt",header = TRUE,sep = "\t")
sample_list <- split(sample_list,sample_list[,1])
for (id in names(sample_list)){
exp_name <- paste0("IP",sample_list[[id]]$IP,"-input",sample_list[[id]]$Input)
ip_sample <- paste0(sample_list[[id]]$IP,".bam")
input_sample <- paste0(sample_list[[id]]$Input,".bam")
exomePeak2(bam_ip =ip_sample, bam_input =input_sample, gff ="ath.gtf", save_dir = getwd(),experiment_name = paste0("exomePeak2_output",exp_name))
}
#notice:ath is the spell in a simplified form of Arabidopsis thaliana
## run exomePeak2.r
nohup Rscript exomePeak2.r &
# Annotation Analysis
This process was already in the RNAmod database, please refer to RNAmod
If you use this analysis pipeline, please cite: Liu, Q. and R.I. Gregory, RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res, 2019. 47(W1): p. W548-W555.
# Citation
If you make use of the data and web-server presented here, please cite our PRMD paper (2023) in addition to the primary data sources.
The PRMD data files can be freely downloaded and used in accordance with the GNU Public License and the license of primary data sources.